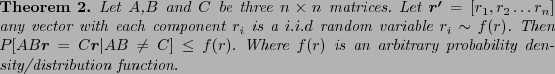De Bruijn graphs recently found applications in
Sequence Assembly(
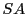
).
problem is close to
Shortest Common super String(
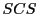
) problem, however
there are some fundamental differences. Given a set of input strings

strings the
problem outputs a string

such that every
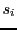
is a sub-string in
and

. On the other hand the input to the
problem
is a set
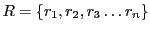
of strings, every

is a randomly chosen
sub-string of a bigger string

- called the
genome. Unfortunately we do not
know what
is, so given
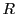
the
problem asks to find the
such that we can
explain every
. Adding to the complexity of
problem
may contain several
repeating regions and the direct appliction of
to solve
problem would
result in collapsing the repeating regions in
- so we cannot directly reduce
the
problem to
. Also the string
is special string coming from the
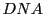
so its double stranded and each
may be originating from the forward or
reverse compliments of
.
![\includegraphics[scale=0.5]{loops_debrujin.eps}](http://vamsi99.users.sourceforge.net/blg/LOOP-2009/img19.png)
A  De Bruijn graph
De Bruijn graph  on a set of strings is a graph whose vertices
correspond to the unique mers (a string of length
on a set of strings is a graph whose vertices
correspond to the unique mers (a string of length 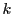 ) from all the
) from all the 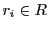 and whose edges correspond to a some
and whose edges correspond to a some  mer in some
mer in some  . Also to consider
the double stranded-ness we also include a reverse compliment of every mer into
the graph . In the following simple example I show how a loop (i.e. both
the forward and reverse compliments overlapping each other by
. Also to consider
the double stranded-ness we also include a reverse compliment of every mer into
the graph . In the following simple example I show how a loop (i.e. both
the forward and reverse compliments overlapping each other by  ) can originate
into . This interesting situation seems to be referred to as hairpin-loop.
) can originate
into . This interesting situation seems to be referred to as hairpin-loop.
![\begin{theorem}
Let $A$,$B$\ and $C$\ be three $n\times n$\ matrices such that $...
...tribution. Then $P[AB\vec{r} = C\vec{r} \vert AB \neq C] \leq 1/2$
\end{theorem}](http://vamsi99.users.sourceforge.net/blg/RAND-2009/img20.png)